How a Metabolic Enzyme Can Trigger Cell Death in Liver Cancer Cells
Oncotarget
October 19, 2023In a new editorial paper, researchers highlight the role of GLS2 in regulating ferroptosis in hepatocellular carcinoma.
—
Hepatocellular carcinoma (HCC) is the most common type of liver cancer. It is particularly challenging to treat because HCC is often diagnosed in late stage and resistant to chemotherapy and radiation. However, advancements in targeted therapies and immunotherapies have opened new avenues for the treatment of this aggressive disease.
In a new editorial paper, researchers Sawako Suzuki, Divya Venkatesh, Tomoaki Tanaka, and Carol Prives from Columbia University highlight the role of a metabolic enzyme known as glutamine synthase 2 (GLS2) in regulating ferroptosis in HCC. Ferroptosis is a form of cell death that involves iron-dependent accumulation of lipid peroxides. On October 19, 2023, their editorial was published in Oncotarget, entitled, “GLS2 shapes ferroptosis in hepatocellular carcinoma.”
GLS2 Promotes Ferroptosis in HCC
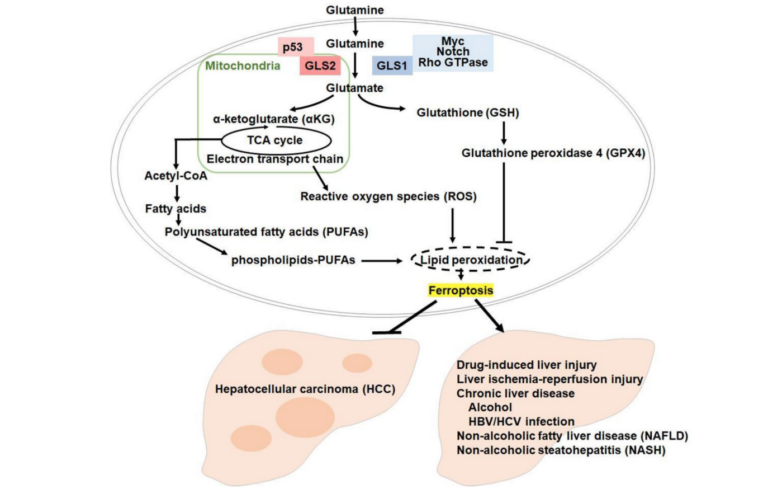
GLS2 is a key enzyme that catalyzes the conversion of glutamine to glutamate, a precursor of alpha-ketoglutarate (αKG), a molecule that participates in several metabolic pathways, such as the tricarboxylic acid (TCA) cycle, redox homeostasis and lipid and amino acid metabolism. GLS2 is also a transcriptional target of the tumor suppressor protein p53, which regulates its expression in response to cellular stress.
In this editorial, the researchers summarize findings from their recent study, which demonstrated that GLS2 is a bona fide tumor suppressor and a regulator of ferroptosis in HCC using mouse models and human cancer cells. The team showed that GLS2 deficiency leads to increased HCC tumorigenesis and resistance to ferroptosis, while GLS2 overexpression reduces tumor growth and sensitizes cancer cells to ferroptosis.
The mechanism by which GLS2 promotes ferroptosis involves its catalytic activity, which facilitates the production of αKG from glutamate. αKG then enhances lipid reactive oxygen species (ROS) generation by inhibiting the activity of glutathione peroxidase 4 (GPX4), an enzyme that protects cells from lipid peroxidation. Thus, GLS2 acts as a metabolic switch that favors ferroptosis by increasing lipid ROS levels.
“Our work has now provided evidence that GLS2 is mainly localized in mitochondria and induces ferroptosis through α-ketoglutarate (αKG), and this occurs specifically under conditions where the levels of GSH [glutathione] or of glutathione peroxidase 4 (GPX4) are suppressed by ferroptosis inducers [7].”
Conclusions
The authors also provided evidence that GLS2-mediated regulation of ferroptosis has clinical relevance for human HCC. They found that GLS2 expression is frequently downregulated in human HCC samples and correlates with poor prognosis. Moreover, they showed that GLS2 expression is associated with sensitivity to erastin, a ferroptosis-inducing agent, in human HCC cell lines.
These results suggest that GLS2 is a potential therapeutic target for HCC and that its modulation could enhance the efficacy of ferroptosis-based therapies. The editorial paper concludes by discussing the challenges and opportunities for further research on the role of GLS2 and ferroptosis in liver disease.
“If indeed GLS2 can promote chemically-induced ferroptosis irrespective of the tissue type, then the drug regimen will need to be tailored such that the liver tissues adjacent to HCC are protected. Taking these concerns into consideration, we hope that our findings will inform future decisions regarding treatment of liver disease.”
Click here to read the full editorial paper in Oncotarget.
—
Oncotarget is an open-access, peer-reviewed journal that has published primarily oncology-focused research papers since 2010. These papers are available to readers (at no cost and free of subscription barriers) in a continuous publishing format at Oncotarget.com. Oncotarget is indexed/archived on MEDLINE / PMC / PubMed.
Click here to subscribe to Oncotarget publication updates.
For media inquiries, please contact media@impactjournals.com.